We are interested in bioinformatics, molecular evolution, and molecular population genetics.
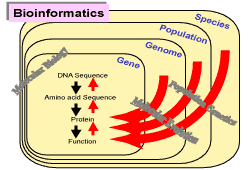 Genome projects are producing almost an infinite amount of molecular data. These genomic sequences
present molecular information constructing current organisms. They are also the results of long
molecular evolution since the origin of life. The genomic sequences are, therefore, filled with
evolutionary footprints. Our interest revolves around mining such information from molecular
sequence data.
Genome projects are producing almost an infinite amount of molecular data. These genomic sequences
present molecular information constructing current organisms. They are also the results of long
molecular evolution since the origin of life. The genomic sequences are, therefore, filled with
evolutionary footprints. Our interest revolves around mining such information from molecular
sequence data.
In order to analyze information gleaned from genomic sequences, it is necessary to understand the rules of biochemistry and molecular biology. Understanding the mechanisms of molecular evolution and population genetics helps us to sift through superficial data and to incorporate another dimension in our study, i.e., time. Developing and incorporating new statistical and computational methods/tools needs to be emphasized to handle a large amount of data efficiently, and to extract biological information from highly noisy data.
Current and Past Projects:
Evaluation and development of next-generation sequencing based methods. With the arrival of next- and third-generation sequencing technologies, it has become possible to acquire the whole genome and transcriptome data rapidly from a variety of organisms. While many new computationl methods are being developed to deal with these new types of data, many challenges remain and thorough performance analysis among the methods is needed. We have been evaluating various methods such as RNA-seq gene expression analysis methods (Zheng and Moriyama 2013) and read mappers (Pavlovikj et al. 2017). We also showed that the quality of genome assembly and annotation significantly affects the outcomes of molecular evolutionary analysis (Gradnigo et al. 2016). For transcriptome assemblers, using simulated benchmark data, we compared the performance of genome-guided and de novo assembly methods and showed that different methods produce different transcriptome assemblies with no single method being apparently the best (Voshall and Moriyama, 2018). Based on the results of the performance analysis, we are currently developing an ensemble method to combine the results from multiple de novo transcriptome assemblers to obtain improved transcriptome assembly.
Mathematical modeling of protein-domain space. Our goals are to establish the complete inventory of protein families within the protein universe and to elucidate their structural and functional relationships. Proteins often share conserved domains. They are the units of protein functions and evolution. By understanding the compositional relationships between the protein space and the domain space, we can gain key information on how proteins acquire functions and how protein functions have evolved within the protein universe. Selective constraints on their sequences maintain protein functions while modularity of domains allows functional innovation. With this assumption, we introduced a game-theoretic method for constructing protein networks (Deng et al. 2013). We further adapted this approach into the framework of multi-objective optimization and developed a new method, MOCASSIN-prot (Keel et al. 2018). While this method can be applied to highly heterogeneous proteome-level classification where phylogenetic analysis is not applicable, it depends on accurate detection of protein domains. We are thus developing more sensitive and accurate domain discovery methods by extending profile hidden Markov models of multi-domain proteins. To evalute the performance of these methods, we are also developing a simulation method for protein family evolution where domains are incorporated as evolutionary units.

Bioinformatics application for western corn rootworm management. (collaboration with Ana Vélez, Hideaki Moriyama, and Blair Siegfried Labs) The western corn rootworm (WCR, Diabrotica virgifera virgifera) is the most serious pest of cultivated maize in North America. Rootworm management has been challenged by this insect's remarkable capacity to evolve resistance to a variety of pest management technologies. To overcome this problem, by combining our knowlege on WCR biology and molecular evolutionary mechanisms of chemoreceptors and other other proteins (Eyun et al. 2014; Rodrigues et al. 2016), we are exploring the use of highly divergent WCR proteins as WCR-specific RNAi targets.
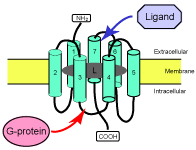
Classification of Seven Transmembrane Receptor (7TMR) superfamily. 7TMRs (as known as G-protein coupled receptor, GPCRs) form a large superfamily of proteins composed of six major classes and more than 30 subfamilies. They are integral membrane proteins characterized by seven membrane-spanning (transmembrane; TM) regions. They are involved with signal transduction across cell membranes. Many medically and pharmacologically important proteins are included in this superfamily (e.g., acetylcholine receptors, dopamine receptors, odorant receptors, etc.). Other than the structural similarity (7 TM regions), sequence similarity among different 7TMR groups is limited, and attempting to find new 7TMRs from new genomic data is often hindered due to such extreme diversity. To overcome this problem, we developed alignment-free approaches to detect weak similarity (Moriyama and Kim, 2003; Moriyama et al. 2006; Strope and Moriyama, 2007; Opiyo and Moriyama, 2007; Moriyama and Opiyo, 2010). In the 7TMRmine Web server, we integrated 14 classifiers including both alignment-based and alignment-free methods (Lu et al. 2009). These methods have been applied to other highly divergent protein families such as the Cytochrome b561 familiy (Opiyo and Moriyama, 2009, 2010).
Development of improved multiple alignment (MSA) methods.
Construction of MSAs is the most fundamental step in almost all bioinformatics and molecular evolutionary analysis.
Development of MSA methods is thus one of the most important and actively researched areas in bioinformatics. This is also one of
the most neglected processes in practice. Users often choose only a single or a handful of MSA method(s) based solely on their
accessibility. In order to change such situations, we have developed SuiteMSA, a visual tool that can be used to generate and
compare MSAs (Anderson et al. 2011a, 2011b). We have also developed a simulation method for biological sequence evolution,
indel-Seq-Gen (iSG) (Strope et al. 2007, 2009). iSG can incorporate many biologically realistic evolutionary events including
insertions and deletions. Simulated sequences can represent a variety of evolutionary scenarios and they are useful for testing MSA
and phylogenetic tree methods. One of our goals is to find a
way to improve multiple alignment of highly divergent protein sequences such as 7TMRs and
 multidomain protein families, and attempt to reconstruct more reliable phylogenies from such divergent sequences.
multidomain protein families, and attempt to reconstruct more reliable phylogenies from such divergent sequences.
Synonymous codon usage bias. Nucleotide substitutions between synonymous codons do not change amino acids. However in many organisms, they do not seem to be neutral to natural selection. Rather they are under a weak selection. Large variations in codon usage bias have been found among genes within the same genome. It can vary also between different regions along the gene, as well as between homologous genes from different species (reviewed in Moriyama, 2003). While this quantity, as well as base composition, has become one of the most routinely obtained information from genomic sequences, it has not been well-understood and has been under-utilized in genomic analyses. One of our goals is to incorporate such information in bioinformatics tools and achieve thorough and multi-dimensional understanding of genomic data.
Project websites:
Publications:
Graduate Student Dissertations and Theses:
Adam Voshall (2018) Consensus ensemble approaches improve de novo transcriptome assemblies. MS thesis, Department of Computer Science and Engineering, University of Nebraska—Lincoln (PDF file)
Catherine Anderson (2017) Selecting the "closest to optimal" multiple sequence alignment using multi-layer perceptron. PhD dissertation, Department of Computer Science and Engineering, University of Nebraska—Lincoln (PDF file)
Julien Gradnigo (2016) Sequencing and comparative analysis of de novo genome assemblies of Streptomyces aureofaciens ATCC 10762. MS thesis, School of Biological Sciences, University of Nebraska—Lincoln (PDF file)
Eric Rodene (2016) Use of clustering techniques for protein domain analysis. MS thesis, Department of Computer Science and Engineering, University of Nebraska—Lincoln (PDF file)
Ling Zhang (2015) Application of linker length and linker length dependency in identification of protein domains. PhD dissertation, Department of Statistics, University of Nebraska—Lincoln (PDF file)
Brittney Hinds (2015) Bioinformatic game theory and its application to cluster multi-domain proteins. PhD dissertation, Department of Mathematics, University of Nebraska—Lincoln (PDF file)
Adam Voshall (2015) Investigating the role of microRNAs in the response to nitrogen deprivation in the green alga Chlamydomonas reinhardtii. PhD dissertation, School of Biological Sciences, University of Nebraska—Lincoln (PDF file)
Neethu Shah (2013) Clustering and classification of multi-domain proteins. MS thesis, Department of Computer Science and Engineering, University of Nebraska—Lincoln (PDF file)
Seong-il Eyun (2013) The origin and molecular evolution of two multigene families: G-protein coupled receptors and glycoside hydrolase families. PhD dissertation, School of Biological Sciences, University of Nebraska—Lincoln (PDF file)
Ximeng Zheng (2012) Comparative studies of differential gene calling methods for RNA-Seq data. MS thesis, School of Biological Sciences, University of Nebraska—Lincoln (PDF file)
Pooja K. Strope (2011) Functional classification of divergent protein sequences and molecular evolution of multi-domain proteins. PhD dissertation, School of Biological Sciences, University of Nebraska—Lincoln (PDF file)
Jason C. Macrander (2010) Microsatellite development, population structure and demographic histories for two species of Amazonian peacock bass Cichla temensis and Cichla monoculus (Perciformes: Cichlidae). MS thesis, School of Biological Sciences, University of Nebraska—Lincoln (PDF file)
Cory L. Strope (2009) Evaluating indels as characters of biological informativeness. PhD dissertation, Department of Computer Science and Engineering, University of Nebraska—Lincoln (PDF file: 28.4MB)
Stephen O. Opiyo (2007) Protein family classification using multivariate methods. PhD dissertation, Department of Agronomy, University of Nebraska—Lincoln (PDF file)
Chendhore S. Veerappan (2007) Molecular evolution of SET-domain protein families in eukaryotes. MS thesis, School of Biological Sciences, University of Nebraska—Lincoln (PDF file)
Mamta Bajaj (2007) Structural analysis of deoxyuridine triphosphatase from Arabidopsis thaliana. MS thesis, School of Biological Sciences, University of Nebraska—Lincoln (PDF file)
Mamta Bajaj (2005) Development of a prediction method for amphipathic alpha-helices from protein primary structure. MS thesis, Department of Computer Science and Engineering, University of Nebraska—Lincoln (PDF file)
Pooja Khati (2004) Comparative analysis of protein classification methods. MS thesis, Department of Computer Science and Engineering, University of Nebraska—Lincoln (PDF file)
Skanth Ganesan (2004) Comparative analysis of gene prediction methods and development of a fungal genome database system. MS thesis, Department of Computer Science and Engineering, University of Nebraska—Lincoln (PDF file)
Zhifang Wang (2002) Development of a hierarchical protein classification tool. MS project report, Department of Computer Science and Engineering, University of Nebraska—Lincoln (PDF file)

(updated: June 20, 2018)