|
— Impatient users: go to How It Works to start your analysis now! —
Introduction
Protein classification methods:
Computational methods of predicting protein functions rely on detecting similarities among proteins.
A variety of protein classification methods have been developed and used for this purpose.
The majority of protein classification methods are alignment-based. These methods rely on
multiple alignments to build various forms of models (e.g,. regular expression patterns, protein fingerprints,
profile hidden Markov models). However, generating reliable multiple alignments becomes increasingly
difficult when more divergent protein sequences are to be incorporated. Another disadvantage shared
by these multiple alignment-based methods is that their models are built only from "positive samples"
(protein sequences of interests), and information from "negative samples" (unrelated protein sequences)
is not directly incorporated. Since subsequently found proteins are classified based on these models,
possible initial sampling bias is kept and possibly reinforced.
Alignment-free methods:
Alignment-free protein classification methods can overcome these problems. Instead
of using alignments, these methods extract various descriptors from each sequence (e.g.,
amino acid composition, dipeptide frequencies, physico-chemical properties). Using pattern recognition
or multivariate statistical methods, similarities among proteins can be evaluated. One of the
disadvantages of such alignment-free methods is relatively high false positive rates. Refer to
our publications below for more detailed discussion on these issues.
Hierarchical classification:
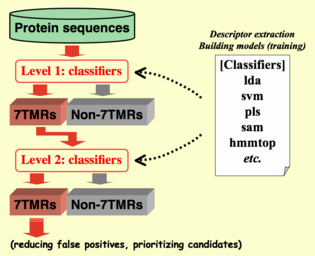 Although alignment-free methods can be more sensitive to difficult-to-detect
remote similarities, they have higher rates of false positives. Alignment-based methods are
more specific (conservative) but less sensitive to remote similarities. In order to perform thorough
mining, we need to take advantages of both types of methods. In order to achieve this, we have
integrated multiple protein classification methods, including both of alignment-based and
alignment-free, in our hierarchical classification system. The power of this approach is
shown in our recent study for mining 7 transmembrane receptor (7TMR) candidates from
the Arabidopsis thaliana genome. As illustrated in the right figure,
multiple sets of classifiers are used as hierarchically ordered filters to extract 7TMR candidates.
By using more sensitive methods earlier and more specific methods later, we
reduced the number of false positives and obtained prioritized target protein sets for further confirmation.
More details on this mining process are described in Moriyama et al. (2006; also in the
accompanying supplementary material web site). Although alignment-free methods can be more sensitive to difficult-to-detect
remote similarities, they have higher rates of false positives. Alignment-based methods are
more specific (conservative) but less sensitive to remote similarities. In order to perform thorough
mining, we need to take advantages of both types of methods. In order to achieve this, we have
integrated multiple protein classification methods, including both of alignment-based and
alignment-free, in our hierarchical classification system. The power of this approach is
shown in our recent study for mining 7 transmembrane receptor (7TMR) candidates from
the Arabidopsis thaliana genome. As illustrated in the right figure,
multiple sets of classifiers are used as hierarchically ordered filters to extract 7TMR candidates.
By using more sensitive methods earlier and more specific methods later, we
reduced the number of false positives and obtained prioritized target protein sets for further confirmation.
More details on this mining process are described in Moriyama et al. (2006; also in the
accompanying supplementary material web site).
Using this web application, users can build their own hierarchical classification systems to examine/mine
extremely divergent 7TMR proteins. In the Home page, users can explore many pre-processed genomes using
this tool. Users can also submit their own sequences for analysis. The dataset and methods published in
Moriyama et al. (2006) is available indenpendently from this link.
Read Methods for description of classifiers included in this application.
Read How It Works to start your analysis.
Currently up to 5MB of user sequences can be submitted for the analysis at once. For a large scale analysis, please
contact the Principle Investigator listed below.
Future plans:
We plan to add SVM-pairwise and other classification as well as transmembrane
prediction methods. More genomes will be added. Our ultimate goal is to extend our classification
system to many other protein families, and establish a proteome classification system.
— Related publications —
Johnston, C. A., Temple, B. R., Chen, J.-G., Gao, Y., Moriyama, E. N., Jones, A. M.,
Siderovski, D. P. and Willard, F. S. (2007) Comment on "A G protein-coupled receptor is a
plasma membrane receptor for the plant hormone abscisic acid".
Science 318: 914.
Kim, J., Moriyama, E. N., Warr, C. G., Clyne, P. J. and Carlson, J. R. (2000)
Identification of novel multi-transmembrane proteins from genomic databases using quasi-periodic structural properties.
Bioinformatics 16: 767-775.
Lu, G., Wang, Z., Jones, A. M. and Moriyama, E. N. (2009)
7TMRmine: A Web server for hierarchical mining of 7TMR proteins.
BMC Genomics 10: 275.
Moriyama, E. N. and Kim, J. (2005)
Protein family classification with discriminant function analysis. Pp. 121-132 in: J.P. Gustafson, R. Shoemaker,
and J.W. Snape, (Eds.), Genome Exploitation: Data Mining the Genome, Springer, New York.
(pdf)
Moriyama, E. N., Strope, P. K., Opiyo, S. O., Chen, Z. and Jones, A. M. (2006)
Mining the Arabidopsis thaliana genome for highly-divergent seven transmembrane receptors.
Genome Biology 7: R96.
— Supplementary Materials —
Opiyo, S. O. and Moriyama, E. N. (2007)
Protein family classification with partial least squares.
J Proteome Res 6: 846-853.
Opiyo, S. O. and Moriyama, E. N. (2009)
Mining the Arabidopsis and rice genomes for cyclophilin protein families.
Int. J. Bioinformatics Research and Applications 5: 295-309.
Opiyo, S. O. and Moriyama, E. N. (2010)
Mining Cytochrome b561 from plant genomes.
Int. J. Bioinformatics Research and Applications 6: 209-221.
Strope, P. K. and Moriyama, E. N. (2007)
Simple alignment-free methods for protein classification: a case study from G-protein coupled
receptors. Genomics 89: 602-612.
— Development team (current and past) —
Univeristy of Nebraksa-Lincoln
- Etsuko N. Moriyama (Principal Investigator, Associate Professor, School of Biological Sciences & Center for Plant Science Innovation)
- Pooja K. Strope (PhD student, Biological Sciences)
- Stephen O. Opiyo (Former PhD student, Agronomy, currently Postdoctral Associate)
- Qiaomei Zhang (Former web programmer)
- Zhifang Wang (Former MS student, Computer Science)
University of Nebraska-Omaha
- Guoqing Lu (Assistant Professor, Departments of Biology and Computer Science)
— Collaborator —
Univeristy of North Carolina at Chapel Hill
- Alan M. Jones (Professor, Departments of Biology & Pharmacology)
— Grant support —
National Library of Medicine/NIH R01LM009219 and 3R01LM009219-02W1

|
